
The Hamilton laboratory is organized into themes converging upon understanding how gastrointestinal epithelial cells function in human health and disease. |
How do RNA binding proteins regulate epithelial cell fate and function in the gastrointestinal tract?
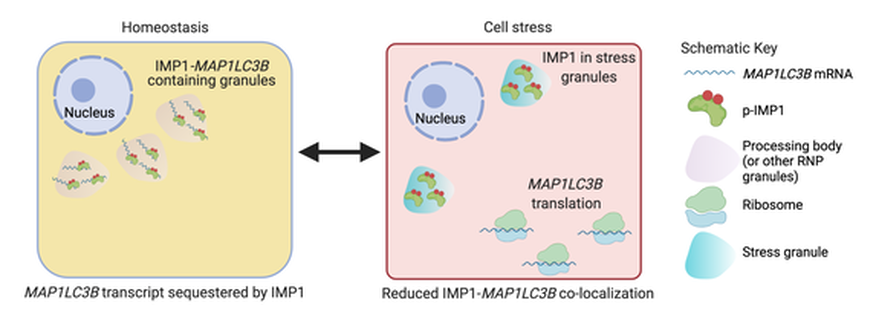
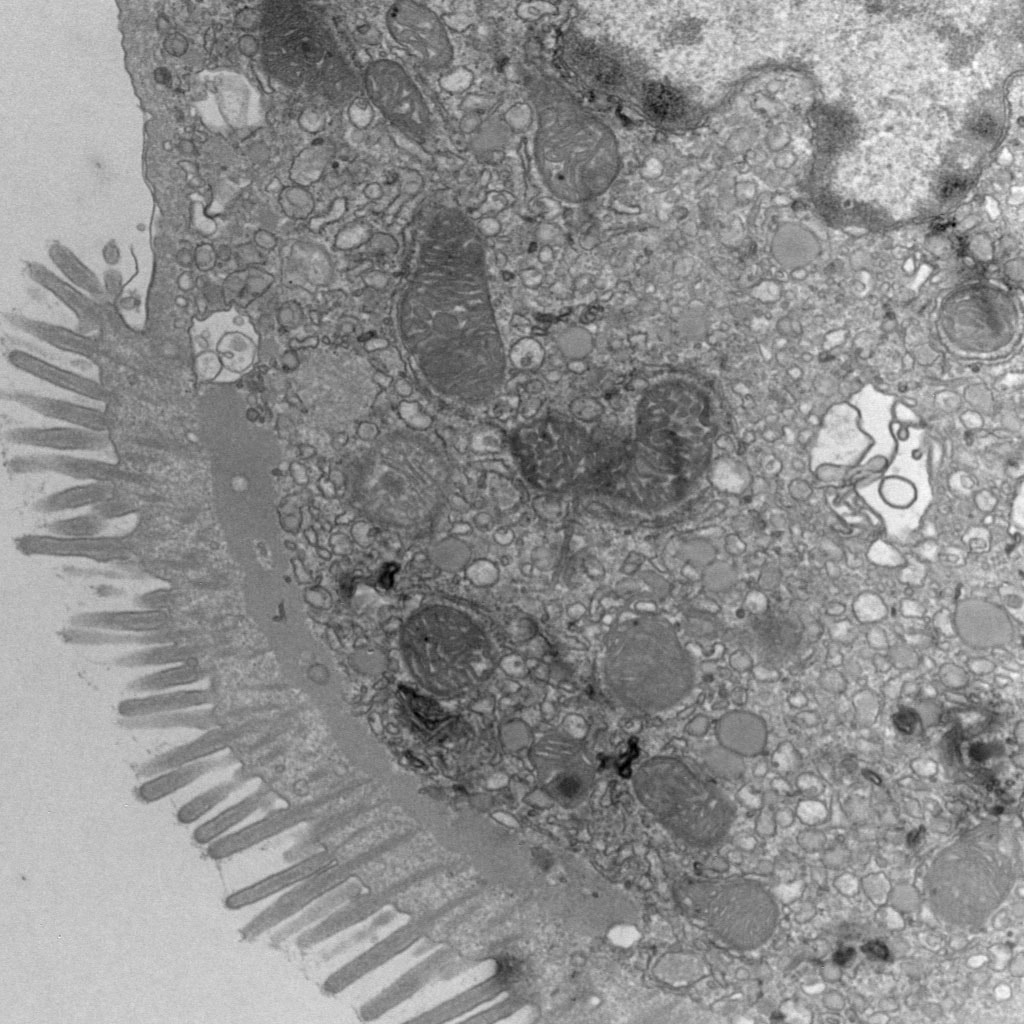
Gene expression regulation can happen at multiple levels. We aim to understand how post-transcriptional gene regulation (i.e. mechanisms regulating RNA -> protein) directs the cell to respond and adapt to changes in the environment. In this context, we are currently focused on post-transcriptional regulation of the autophagy pathway- a pathway with multi-faceted and incompletely understood roles in gastrointestinal epithelial cell subtypes. Understanding post-transcriptional regulation of key cellular pathways (such as autophagy) in the context of disease allows us to better define how normal cells behave and will provide a foundation for developing new therapies. Our recent work identified a new molecular connection between IMP1 and autophagy transcript MAP1LC3B. We found that IMP1 deletion, or mutation of a key IMP1 phosphorylation site, is associated with increased MAP1LC3B protein levels, suggesting that IMP1 can suppress MAP1LC3B translation at homeostasis. This new finding is important because it increases our understanding of how autophagy may be regulated in cells where IMP1 is expressed- namely intestinal stem cells and Paneth cells. Multiple ongoing studies are seeking to understand how IMP1 expression in intestinal stem cells and Paneth cells may differentially regulate autophagy in inflammatory bowel disease and whether this process is dependent upon IMP1's role as a reader of m6A-modified mRNAs.
How does the autophagy pathway contribute to epithelial regeneration?
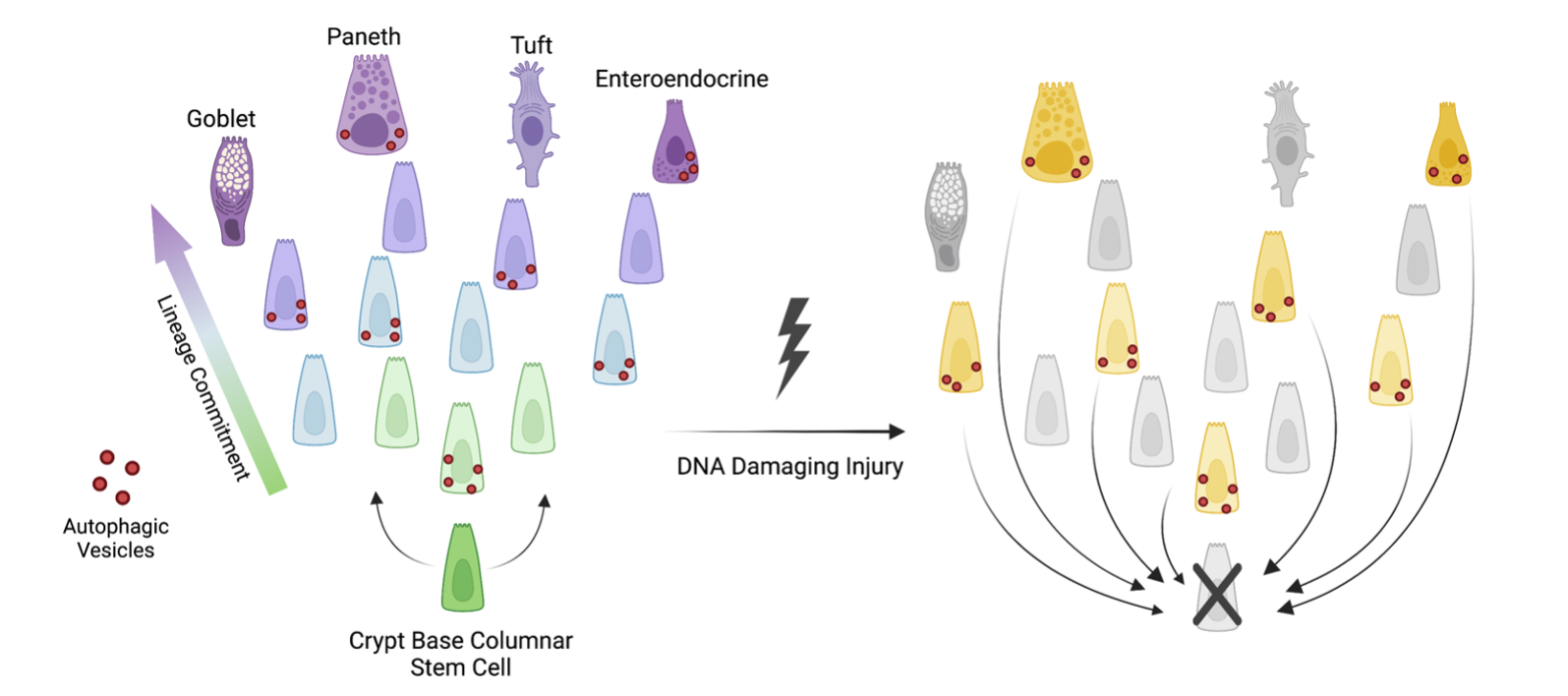
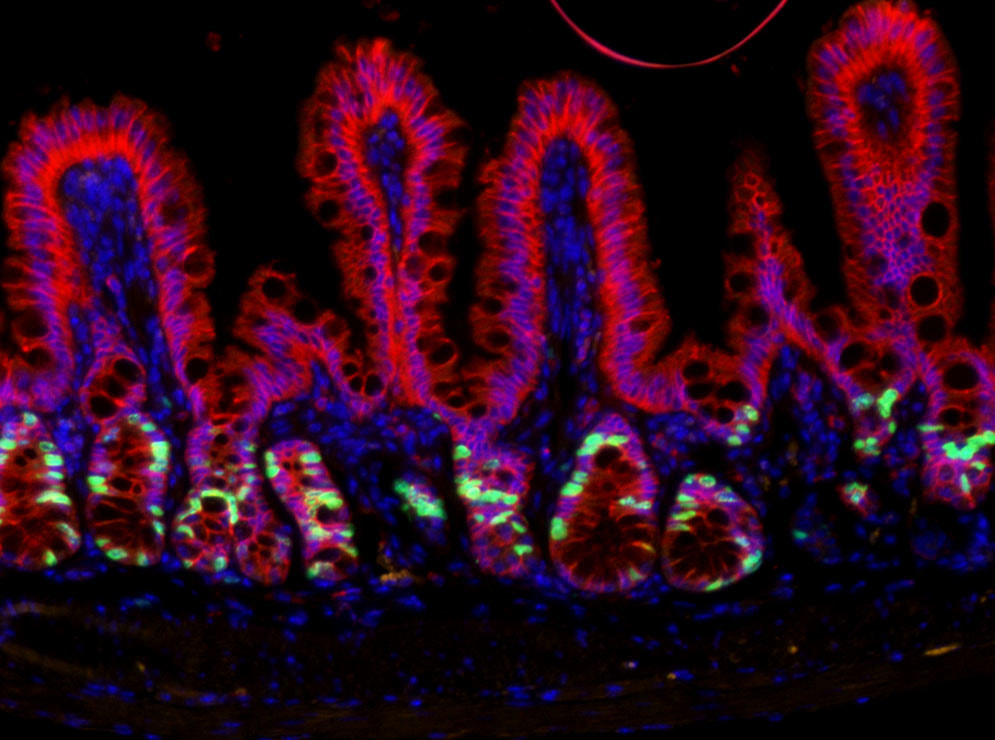
Our studies evaluating post-transcriptional gene regulation of the autophagy pathway led us to wonder how different cells of the epithelial barrier- namely intestinal stem cells- might utilize autophagy to facilitate epithelial barrier integrity. Autophagy deletion in mice increases susceptibility to inflammatory injury and infection. In addition, autophagy deletion renders mice less able to regenerate their epithelial barrier after radiation injury. Finally, genome wides association studies (GWAS) identified multiple autophagy gene risk alleles in patients with inflammatory bowel disease, suggesting that defective autophagy may contribute to human disease pathogenesis. Our emerging studies find that non-stem cells, including cells of the secretory lineage, that have high autophagy activity can act as facultative stem cells. We also found that blocking autophagy seems to inhibit the stem plasticity of non-stem cells. Finally, we find that cells with high autophagy are less likely to exhibit DNA damage following radiation. Our findings support the hypothesis that intestinal epithelial autophagy has a dual role in tissue regeneration: 1) to promote plasticity of non-stem cells to allow them to help regenerate the tissue after injury, and 2) to protect cells from DNA damage.
Do epithelial stem cells in patients with inflammatory bowel disease act differently than healthy patients?
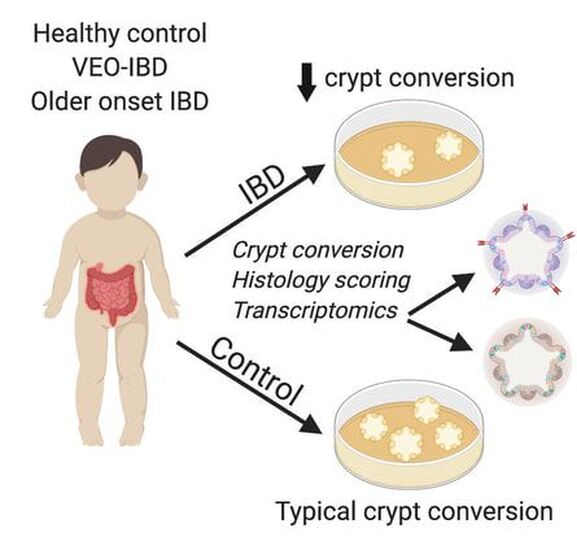
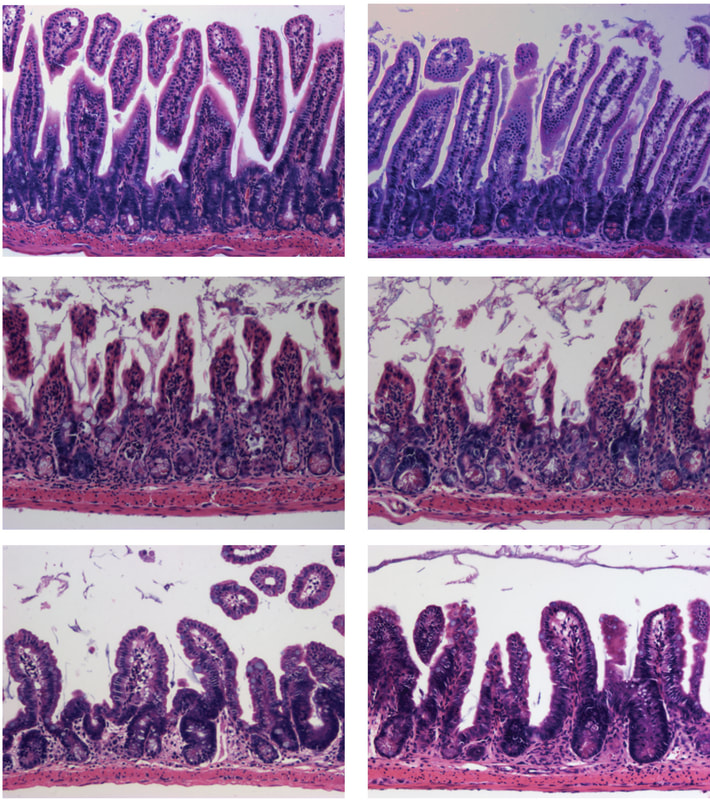
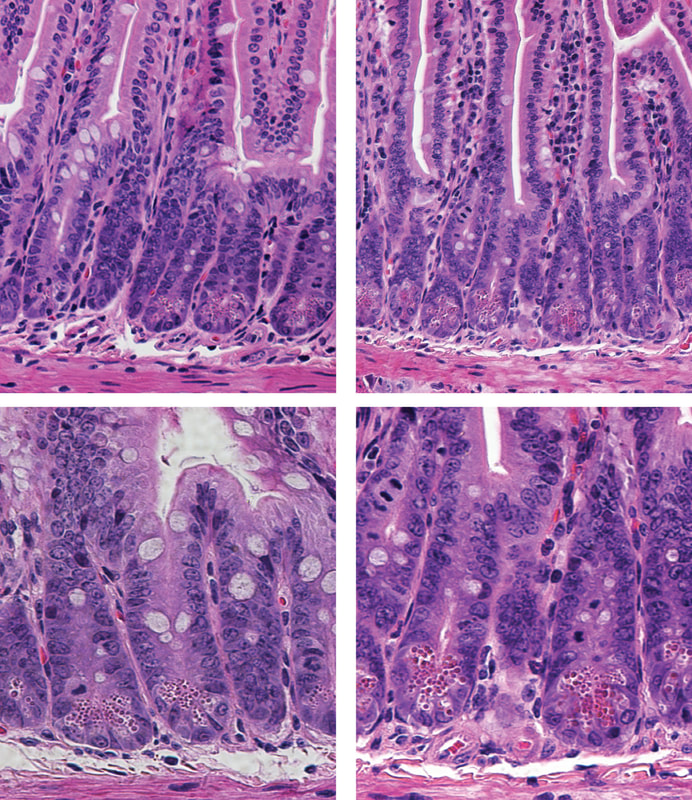
Our laboratory uses 3D organoids (enteroids and colonoids) from patients with inflammatory bowel disease and compares them to healthy patients to understand whether intestinal epithelial stem cells behave differently in health versus chronic disease. We are using enteroid/colonoid technology to understand multiple areas, including: 1) how epithelial stem cells from different patients with IBD survive, grow, and self-renew in culture; 2) how genes traditionally associated with immune cells function in epithelial stem cells and potentially contribute to IBD pathogenesis. These efforts are supported by the Children's Hospital of Philadelphia Gastrointestinal Epithelium Modeling Program (Dr. Hamilton is Co-Director) and the Helmsley Charitable Trust's Gut Cell Atlas initiative.
|
|

|
|